
Em outubro de 2018, a ANVISA publicou o Guia nº 08, na qual contempla o entendimento da Agência sobre as práticas mais reconhecidas com relação a procedimentos, rotinas e métodos de investigação de resultados fora de especificação, adequados ao cumprimento do requisito técnico (Art. 292, seção I da RDC 17 de 2010).
Desde a publicação da RDC 17 (abril/2010) a ANVISA vem cobrando das indústrias a respeito do tema, resultado fora de especificação (FDE), ou mais conhecido como OOS (Out of specification), porém, pouco as empresas sabem sobre o tema. As indústrias multinacionais acabaram saindo na frente, pois seguem os Guias do FDA, mas as indústrias nacionais tiveram que se ajustar para cumprir o requisito regulatório, mesmo que sem muita informação da Agência Nacional.
Quais as orientações mais importantes que o Guia nos traz?
- Como investigar o evento;
- As responsabilidades do pessoal do laboratório e de outros departamentos (garantia da qualidade, produção, pesquisa e desenvolvimento, engenharia e manutenção), quando aplicável;
- A definição de fases da investigação;
- Os testes e amostragens adicionais que podem ser necessários;
- A extrapolação das investigações para fora das instalações laboratoriais;
- Como fazer a avaliação final dos resultados.

Qual é a aplicação do FDE – Resultado Fora de Especificação?
Sua aplicabilidade vai além da análise de produto terminando, inclui também:
- Testes para liberação de matérias-primas, materiais de embalagem, intermediários, materiais de partida, produtos a granel e terminados de medicamentos, insumos farmacêuticos ativos (IFA), e excipientes farmacêuticos;
- Testes de controle em processo – se utilizados para cálculo de rendimento ou para decisão sobre o lote, exceto resultados fora de especificação obtidos durante o ajuste de equipamentos e dispositivos;
- Resultados de estudos de estabilidade de produtos terminados de medicamentos, insumos farmacêuticos ativos (IFA) e excipientes farmacêuticos; e
- Resultados fora de especificação em amostras de retenção de lotes (testados em decorrência de investigação).
Mas o que é FDE – Resultado Fora de Especificação?
Um resultado fora de especificação é quando se obtém um resultado acima ou abaixo da especificação informada no registro do produto.
De acordo com este Guia, este resultado pode ser obtido durante análise laboratorial e/ou também durante o controle em processo do produto.
Os produtos fora das especificações não devem ser liberados para a comercialização ou uso.
Agora que já sabemos o que é um resultado fora de especificação, o que fazer quando ele aparecer?
De acordo com a legislação vigente, quando um resultado FDE é obtido, o mesmo deve ser imediatamente investigado para determinar a causa raiz, ações corretivas e preventivas, por meio de procedimento aprovado.
Mas o que é imediatamente??? De acordo com as práticas de mercado, o usual é a abertura do formulário de investigação em até 24 h.
Vale lembrar! Mesmo que a empresa adote uma política de sempre rejeitar o lote com resultado FDE, a abertura de investigação é requerida, para avaliar se o resultado está associado a outros lotes do mesmo produto ou mesmo de produtos diferentes.
A rejeição de um lote não justifica a NÃO abertura de investigação.
Vamos ao passo a passo (fases) de uma boa investigação
A investigação deve ser iniciada imediatamente a detecção do resultado FDE, a mesma deve ser aprofundada, imparcial, bem documentada e com embasamento técnico.

Quando detectado um resultado FDE, devem ser preservados as soluções, os reagentes e os materiais (por exemplo, vidrarias, filtros etc), os registros da preparação destes compostos, dos instrumentos utilizados e dos analistas envolvidos com a análise, até o final da investigação.
De acordo com o Guia nº 8, figura 1, a investigação é dividida em fases.
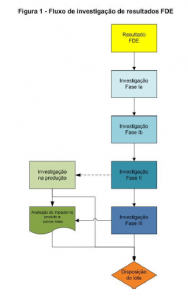
Fase I
Fase de avaliação da exatidão dos resultados, esta fase pode ser dividida em fase Ia e Ib.
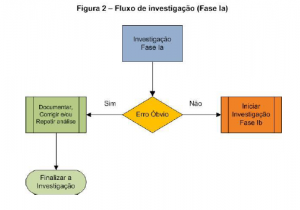
Fase Ia
Nesta fase a investigação fica sob a responsabilidade do analista, é o momento de avaliar a ocorrência de erros óbvio, tais como:
- Queda de energia;
- Falha no equipamento;
- Outros erros detectados pelo analista antes da geração dos dados;
Nesta fase, também é possível verificar erros na execução dos testes, tais como:
- Derramamento de amostras;
- Transferência incompleta de amostras;
- Crescimento de microrganismos em placa de Petri armazenadas fora de locais e condições apropriados;
- Falta de controles positivos ou negativos;
Também faz parte desta fase, a verificação de erros detectados na revisão do analista ou supervisor do laboratório, como por exemplo, a revisão dos cálculos e parâmetros incorretos utilizados na análise.
Todos os erros confirmados devem ser registrados/documentados para manter a rastreabilidade da análise.
Nesta fase, o analista pode interromper a continuidade de análise, a qual tenha detectado um erro óbvio. Na maioria das empresas, a interrupção da análise deve ser autorizada pelo supervisor ou cargo equivalente, e está autorização é bem visto pelas agências reguladoras.
Se não for confirmada a causa do erro, em nenhuma das condições acima, a investigação deve continuar, seguir para fase Ib.
Fase Ib
Nesta fase, precisa de um trabalho conjunto do analista e o supervisor. Deve-se focar nos dados brutos, tais como:
- Nas revisões de dados sobre os equipamentos;
- Materiais (vidrarias, filtros, reagentes);
- Procedimentos;
- Métodos de análises;
Para nortear a investigação, um check list pode ser utilizado, e os itens mínimos devem ser verificados:
- O método analítico executado é a versão aprovada/vigente;
- A validação ou adequabilidade do método analítico executado;
- A amostragem foi realizada conforme procedimento aprovado/vigente. Se registro está disponível, se foi armazenado e transportado de forma correta;
- A integridade das amostras relacionada à estanqueidade do fechamento dos recipientes e ao seu armazenamento temporário;
- Os registros de preparação de meio de cultura e reagentes;
- Os prazos de validade de solução, meios de cultura e reagentes;
- A quantidade de passagens/gerações e a rastreabilidade das cepas de microrganismos padrões;
- Os dados brutos encontrados em todas as etapas da análise, por exemplo, cromatogramas e espectrogramas. A verificação deve buscar a identificação de dados anormais ou suspeitos;
- Os cálculos derivados dos dados brutos e a integridade dos dados. Deve-se incluir nessa verificação a ausência de alterações indevidas nos dados registrados pelos sistemas automatizados (integridade dos dados);
- As calibrações dos instrumentos, seus registros e se os procedimentos de calibração foram realizados apropriadamente;
- As qualificações e os registros de uso de equipamentos utilizados;
- Os livros de registros dos equipamentos e confirmação do desempenho e o uso dos instrumentos designados no procedimento de análise;
- A adequabilidade do sistema;
- O treinamento e a qualificação do analista no método;
- O desempenho do método, ou seja, a concordância do resultado encontrado com o resultado esperado, com base nos dados de validação e dados históricos;
- Os dados de identificação e de qualidade dos padrões de referência (e sua caracterização quando aplicável), padrões de trabalho, soluções reagentes, solventes e outras substâncias utilizadas. A verificação deve buscar dados da validade, especificações, aparência, condições de armazenamento e qualquer outra de suas especificações de controle de qualidade (por exemplo: pré-tratamento como dessecação, correção de teor etc.);
- Limpeza e armazenagem correta dos recipientes, vidrarias e utensílios utilizados na amostragem e na análise;
- Indícios de contaminação da amostra. Por exemplo: a amostra permaneceu aberta ou abandonada; o sistema de insuflamento e exaustão são adequados e estavam funcionando corretamente no momento da amostragem e se houve compartilhamento de utensílios;
- Histórico de problemas relacionados ao ensaio em questão;
- Desvios das condições ambientais relacionadas à temperatura, umidade ou incidência luminosa durante o ensaio;
- Verificação dos dados de lotes que estavam sendo analisados em conjunto; e
- Outras atividades que ocorreram durante o teste e que poderiam interferir no resultado.
Condições adicionais devem ser consideradas e investigadas caso o teste fora da especificação seja microbiológico. Assim, deve-se verificar:
- Se a aparência do meio de cultura nas placas amostrais utilizadas está de acordo com o esperado;
- A localização ou disposição das colônias nas placas e nas áreas de contato amostradas;
- Se o meio de cultura está uniforme e íntegro sobre a placa ou há rachaduras ou outros sinais de degradação;
- A ocorrência de contaminação em outras amostras ou em outros testes realizados na sequência do teste que disparou o evento investigado. Deve-se considerar inclusive os resultados de monitoramento ambiental no período considerado;
- Se os controles negativos e positivos estão de acordo com o esperado;
- Se os meios de culturas e/ou reagentes utilizados estavam corretos e a sua armazenagem antes do uso;
- A integridade do recipiente contendo a amostra;
- As condições de armazenamento da amostra desde a amostragem até o uso no teste;
- Se o tempo entre a amostragem e a realização do teste estava dentro do tempo suportado pelos estudos prévios;
- Se as condições de incubação foram satisfatórias; e
- Se o microrganismo isolado e identificado, motivo do resultado fora de especificação, pode auxiliar na investigação da causa raiz.
Nesta fase o analista deve ser entrevistado para confirmar o seu conhecimento no método executado, também identificar erros que o mesmo deveria ter informado ao supervisor previamente.
Sempre que possível, evidencie as amostras e todo o material com fotografias.
Atenção: erros laboratoriais evidencia problemas de treinamento inadequado, à manutenção precária e calibração imprópria de instrumentos.
Caso a investigação conclua um erro no laboratório, os resultados FDE devem ser invalidados, e as análises repetidas. Caso não haja evidência clara de erro laboratorial, a investigação deve prosseguir para próxima fase.



{kind=link}
{kind=link}
{kind=link}